bands-inspect¶
This is a tool to create, compare and plot bandstructures of materials.
Tutorial¶
The following tutorial illustrates some examples of the most important functionality in the bands-inspect library.
Defining k-points¶
The bands-inspect code defines different ways of defining a set of k-points:
Explicit k-points¶
First, k-points can be given explicitly as a nested list:
In [1]: import bands_inspect as bi
...: import numpy as np
...:
In [2]: kpts_explicit = bi.kpoints.KpointsExplicit(
...: [[0, x, x] for x in np.linspace(0, 1, 6)]
...: )
...:
Out[2]:
namespace(kpoints=array([[0. , 0. , 0. ],
[0. , 0.2, 0.2],
[0. , 0.4, 0.4],
[0. , 0.6, 0.6],
[0. , 0.8, 0.8],
[0. , 1. , 1. ]]))
K-point mesh¶
Second, an even mesh of k-points can be generated by giving the size of the mesh. A constant offset for the mesh points can also be set:
In [3]: kpts_mesh = bi.kpoints.KpointsMesh([2, 3, 2])
Out[3]: namespace(mesh=(2, 3, 2), offset=array([0, 0, 0]))
In [4]: kpts_mesh_offset = bi.kpoints.KpointsMesh([2, 3, 2], offset=[0.1, 0, -0.1])
Out[4]: namespace(mesh=(2, 3, 2), offset=array([ 0.1, 0. , -0.1]))
To check the generated k-points, all such k-point classes have a property kpoints_explicit which contains the explicit list of points:
In [5]: kpts_mesh.kpoints_explicit
Out[5]:
array([[0. , 0. , 0. ],
[0. , 0. , 0.5 ],
[0. , 0.33333333, 0. ],
[0. , 0.33333333, 0.5 ],
[0. , 0.66666667, 0. ],
[0. , 0.66666667, 0.5 ],
[0.5 , 0. , 0. ],
[0.5 , 0. , 0.5 ],
[0.5 , 0.33333333, 0. ],
[0.5 , 0.33333333, 0.5 ],
[0.5 , 0.66666667, 0. ],
[0.5 , 0.66666667, 0.5 ]])
In [6]: kpts_mesh_offset.kpoints_explicit
Out[6]:
array([[0.1 , 0. , 0.9 ],
[0.1 , 0. , 0.4 ],
[0.1 , 0.33333333, 0.9 ],
[0.1 , 0.33333333, 0.4 ],
[0.1 , 0.66666667, 0.9 ],
[0.1 , 0.66666667, 0.4 ],
[0.6 , 0. , 0.9 ],
[0.6 , 0. , 0.4 ],
[0.6 , 0.33333333, 0.9 ],
[0.6 , 0.33333333, 0.4 ],
[0.6 , 0.66666667, 0.9 ],
[0.6 , 0.66666667, 0.4 ]])
K-point path¶
Finally, the k-points can be described as a set of special points, and one or more paths going between these paths. The paths are given as a nested list of identifiers, while the points itself are given as a dictionary mapping these identifiers to the corresponding coordinate:
In [7]: kpts_path = bi.kpoints.KpointsPath(
...: paths=[['L', 'X', '$\Gamma$'], ['L', '$\Gamma$']],
...: special_points={
...: '$\Gamma$': [0, 0, 0],
...: 'X': [0, 0.5, 0.5],
...: 'L': [0.5, 0.5, 0.5],
...: }
...: )
...:
Out[7]: <bands_inspect.kpoints._path.KpointsPath at 0x7fa359cde5f8>
In [8]: kpts_path.kpoints_explicit
Out[8]:
array([[5.00000000e-01, 5.00000000e-01, 5.00000000e-01],
[4.99840866e-01, 5.00000000e-01, 5.00000000e-01],
[4.99681731e-01, 5.00000000e-01, 5.00000000e-01],
...,
[1.83789745e-04, 1.83789745e-04, 1.83789745e-04],
[9.18948723e-05, 9.18948723e-05, 9.18948723e-05],
[0.00000000e+00, 0.00000000e+00, 0.00000000e+00]])
Note that this produces k-points which are equally spaced in reduced coordinates, not in the more physical Cartesian coordinates. To remedy this, a unit cell can be given. Furthermore, the maximum distance between any two k-points can be controlled by a parameter kpoint_distance:
In [9]: a = 3.2395
...: kpts_path_with_uc = bi.kpoints.KpointsPath(
...: paths=[['L', 'X', '$\Gamma$'], ['L', '$\Gamma$']],
...: special_points={
...: '$\Gamma$': [0, 0, 0],
...: 'X': [0, 0.5, 0.5],
...: 'L': [0.5, 0.5, 0.5],
...: },
...: kpoint_distance=0.1,
...: unit_cell=[[0, a, a], [a, 0, a], [a, a, 0]]
...: )
...:
Out[9]: <bands_inspect.kpoints._path.KpointsPath at 0x7fa359cdec50>
In [10]: kpts_path_with_uc.kpoints_explicit
Out[10]:
array([[0.5 , 0.5 , 0.5 ],
[0.4375, 0.5 , 0.5 ],
[0.375 , 0.5 , 0.5 ],
[0.3125, 0.5 , 0.5 ],
[0.25 , 0.5 , 0.5 ],
[0.1875, 0.5 , 0.5 ],
[0.125 , 0.5 , 0.5 ],
[0.0625, 0.5 , 0.5 ],
[0. , 0.5 , 0.5 ],
[0. , 0.45 , 0.45 ],
[0. , 0.4 , 0.4 ],
[0. , 0.35 , 0.35 ],
[0. , 0.3 , 0.3 ],
[0. , 0.25 , 0.25 ],
[0. , 0.2 , 0.2 ],
[0. , 0.15 , 0.15 ],
[0. , 0.1 , 0.1 ],
[0. , 0.05 , 0.05 ],
[0. , 0. , 0. ],
[0.5 , 0.5 , 0.5 ],
[0.4375, 0.4375, 0.4375],
[0.375 , 0.375 , 0.375 ],
[0.3125, 0.3125, 0.3125],
[0.25 , 0.25 , 0.25 ],
[0.1875, 0.1875, 0.1875],
[0.125 , 0.125 , 0.125 ],
[0.0625, 0.0625, 0.0625],
[0. , 0. , 0. ]])
Creating eigenvalue objects¶
The data container used in the bands-inspect code is the EigenvalsData class. It contains a list of k-points (either given explicitly or as a k-points object), and a corresponding array of eigenvalues:
In [11]: ev_explicit = bi.eigenvals.EigenvalsData(
....: kpoints=[[0, 0, 0], [0.1, 0.1, 0.1]],
....: eigenvals=[[-0.124, 0.001123, 0.51234], [-0.132, 0.013, 0.412]]
....: )
....:
Out[11]:
namespace(kpoints=namespace(kpoints=array([[0. , 0. , 0. ],
[0.1, 0.1, 0.1]])),
eigenvals=array([[-0.124 , 0.001123, 0.51234 ],
[-0.132 , 0.013 , 0.412 ]]))
When a Python function that calculated the eigenvalues at a given k-point is known, the from_eigenval_function() method can be used to conveniently build such an EigenvalsData object. For example, if we have a tight-binding model from the TBmodels code:
In [12]: import tbmodels
....: model = tbmodels.io.load(SAMPLES_DIR / 'InSb.hdf5');
....:
In [13]: ev_from_function = bi.eigenvals.EigenvalsData.from_eigenval_function(
....: kpoints=kpts_path_with_uc,
....: eigenval_function=model.eigenval
....: );
....:
Plotting¶
Having created the EigenvalsData instance, we can plot the bandstructure with the plot.eigenvals() function:
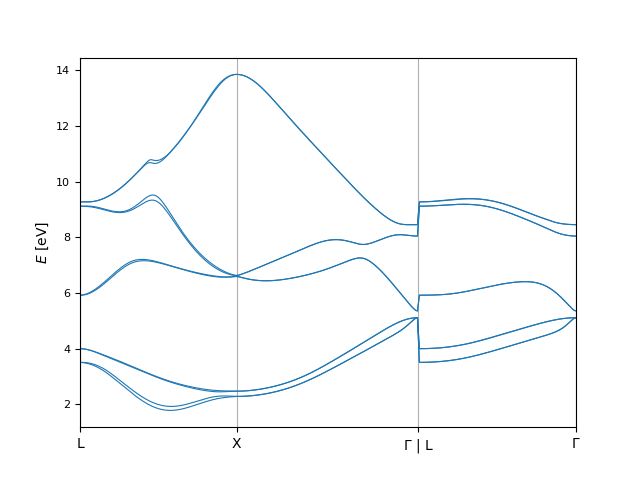
In [14]: bi.plot.eigenvals(ev_from_function);
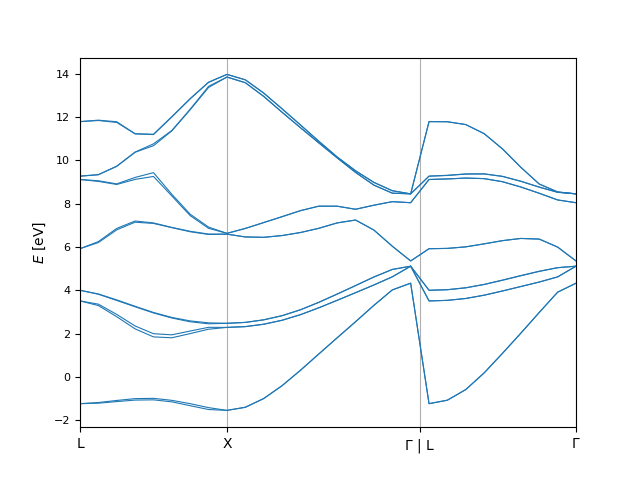
Of course, we can create a finer version of this plot by repeating the same procedure with a smaller kpoint_distance:
In [15]: kpt_fine = bi.kpoints.KpointsPath(
....: paths=[['L', 'X', '$\Gamma$'], ['L', '$\Gamma$']],
....: special_points={
....: '$\Gamma$': [0, 0, 0],
....: 'X': [0, 0.5, 0.5],
....: 'L': [0.5, 0.5, 0.5],
....: },
....: kpoint_distance=0.01,
....: unit_cell=[[0, a, a], [a, 0, a], [a, a, 0]]
....: )
....: ev_fine = bi.eigenvals.EigenvalsData.from_eigenval_function(
....: kpoints=kpt_fine,
....: eigenval_function=model.eigenval
....: )
....: bi.plot.eigenvals(ev_fine);
....:
Saving objects¶
K-point and eigenvalue objects (and combinations of such objects) can be saved in the HDF5 format using the io.save() function, and loaded using io.load():
In [16]: import tempfile
....: with tempfile.NamedTemporaryFile() as tf:
....: bi.io.save([kpt_fine, ev_explicit], tf.name)
....: kpt_res, ev_res = bi.io.load(tf.name)
....:
In [17]: kpt_res
Out[17]: <bands_inspect.kpoints._path.KpointsPath at 0x7fa3582e22b0>
In [18]: ev_res
Out[18]:
namespace(kpoints=namespace(kpoints=array([[0. , 0. , 0. ],
[0.1, 0.1, 0.1]])),
eigenvals=array([[-0.124 , 0.001123, 0.51234 ],
[-0.132 , 0.013 , 0.412 ]]))
Band difference¶
The bands-inspect library also defines functionality for comparing bandstructures. Specifically, it allows for computing the difference between two sets of eigenvalues with equal k-points. This is implemented in the compare.difference.calculate() function. By default, it computes the average difference
where \(i\) is the band index.
In [19]: ev_fine_modified = bi.eigenvals.EigenvalsData.from_eigenval_function(
....: kpoints=kpt_fine,
....: eigenval_function=lambda k: model.eigenval(k) * 1.1
....: )
....:
....: bi.compare.difference.calculate(ev_fine, ev_fine_modified)
....:
Out[19]: 0.641595461618997
Since not all energy ranges or k-points are always equally important when comparing band-structures, it is also possible to define weights for the k-points and energies. The computed difference then becomes
The weights are given as functions which take all k-points / energies and return a corresponding array of weights. For the energies, one can choose whether the function is supplied with the average of the two values (if symmetric_eigenval_weights=True) or only the first value. This latter option is useful if many different band structures need to be compared against a single reference, to ensure that the weights are always the same.
For example, let us calculate the difference where only the first \(100\) k-points and energies above \(6~\text{eV}\) are taken into account:
In [20]: def kpt_weight(kpts):
....: res = np.zeros(np.array(kpts).shape[0])
....: res[:100] = 1
....: return res
....:
In [21]: def ev_weight(energy):
....: return np.array(energy > 6, dtype=int)
....:
In [22]: bi.compare.difference.calculate(
....: ev_fine,
....: ev_fine_modified,
....: weight_kpoint=kpt_weight,
....: weight_eigenval=ev_weight,
....: )
....:
Out[22]: 0.9713096623784362
Furthermore, the function used to calculate the average can also be defined by the user. By default, np.average is used.
Alignment¶
A common problem when comparing band structures is that they can be shifted in energy relative to each other. Often this is not a physical effect, but due to e.g. using a different DFT code, or different pseudopotentials.
Here, we manually create such a shifted band structure.
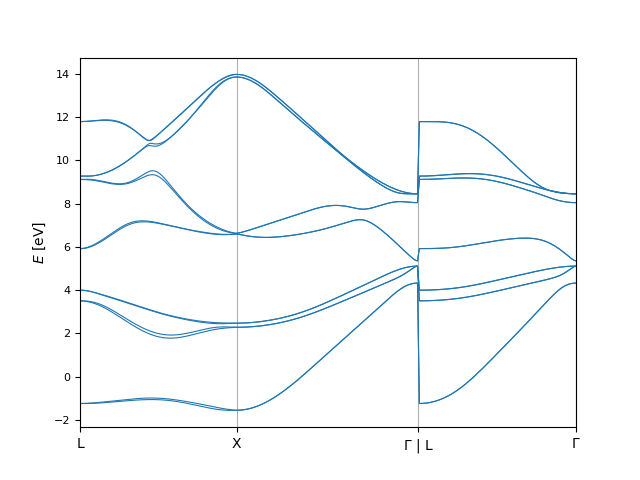
In [23]: ev_shifted = bi.eigenvals.EigenvalsData(
....: kpoints=ev_fine.kpoints,
....: eigenvals=ev_fine.eigenvals + 2.
....: )
....: fig, ax = plt.subplots()
....: bi.plot.eigenvals(
....: ev_fine,
....: ax=ax,
....: plot_options={'color': 'C0', 'linewidth': 0.8}
....: )
....: bi.plot.eigenvals(
....: ev_shifted,
....: ax=ax,
....: plot_options={'color': 'C1', 'linewidth': 0.8}
....: );
....:
The compare.align sub-module can be used to re-align these band structures: It computes the energy shift needed to minimize the difference between the two band structures.
In [24]: align_result = bi.compare.align.calculate(ev_fine, ev_shifted)
....: align_result.shift
....:
Out[24]: -2.0000000020334427
In [25]: align_result.difference
Out[25]: 2.0334427518568397e-09
It also directly returns the aligned band structures:
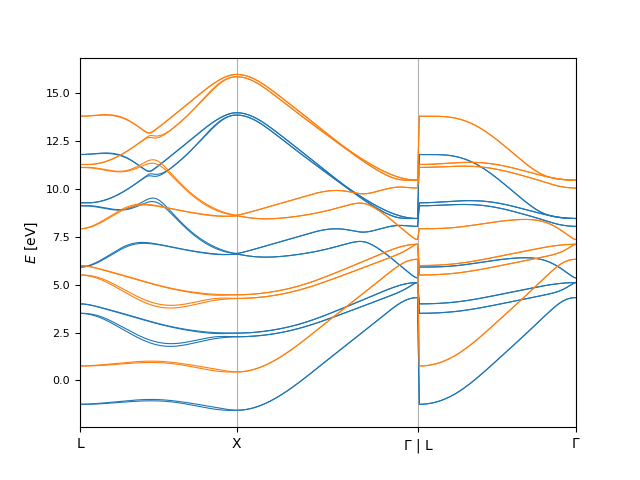
In [26]: fig, ax = plt.subplots()
....: bi.plot.eigenvals(
....: align_result.eigenvals1_shifted,
....: ax=ax,
....: plot_options={'color': 'C0', 'linewidth': 0.8}
....: )
....: bi.plot.eigenvals(
....: align_result.eigenvals2_shifted,
....: ax=ax,
....: plot_options={'color': 'C1', 'linewidth': 0.8, 'linestyle': '--'}
....: );
....:
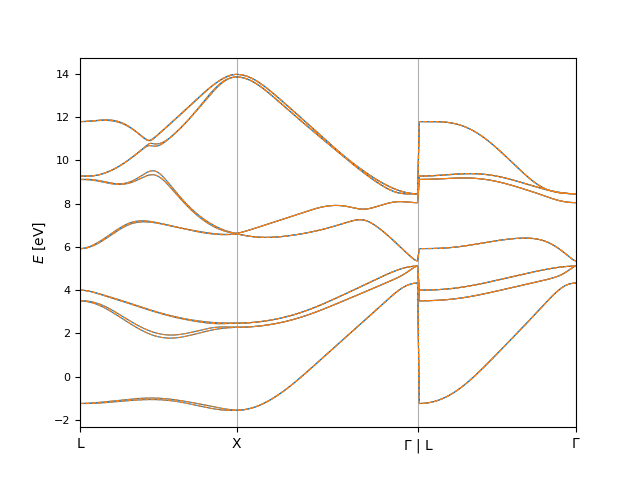
Note here that only the second set of eigenvalues was shifted. This can be changed with the symmetric_shift keyword argument to compare.align.calculate().
Shifting in a non-symmetric way is useful when e.g. comparing multiple band structures to a given reference, to keep the reference unchanged.
Slicing bands¶
Often, band structures with different number of bands need to be compared. To do this, the bands need to first be reduced such that their band indices match. This can be done using the slice_bands() method of EigenvalsData objects. In the following example, we reduce the previously seen bandstructure of InSb by cutting the two lowest and highest bands.
In [27]: ev_sliced = ev_fine.slice_bands(range(2, 12))
....: bi.plot.eigenvals(ev_sliced);
....: